2026
23
Qiang Wang, Tan Li, Pengru Huang, Qi Yu, Kun Fu, Shibo Xi, Xiaocang Han, Jingcong Hu, Xiaoxu Zhao, Haipei Shao, Ming Lin, Yang Meng, Jinxing Chen, Jiali Li, Caozheng Diao, Xiao Hai, Yulin Wang, Xingjie Fu, Jialu Sun, Kostya S. Novoselov, Richard Y. Liu, Jun Li, and Jiong Lu
Geminal Atom Catalysts with Minimized d-Orbital Holes Enable β-Elimination-Resistant C(sp2)–C(sp3) Cross-Coupling
-
Heterogeneous C(sp2)–C(sp3) Suzuki–Miyaura coupling offers an attractive route for the large-scale and sustainable synthesis of structurally complex and pharmaceutically relevant molecules that are otherwise difficult to access. However, the low reactivity of unactivated alkyl electrophiles and the intrinsic instability of alkyl metal intermediates, particularly their propensity for β-hydride elimination, render selective C(sp2)–C(sp3) bond formation exceptionally challenging. Here, we integrate high-throughput density functional theory (DFT) screening with quantum-chemical calculations to identify Cu-based geminal-atom catalysts as optimal candidates and uncover the critical role of d-orbital holes that promote agostic interactions, leading to undesired β-hydride elimination. Guided by these insights, we develope a d-orbital hole passivation strategy to fabricate a class of high-fidelity Cu-based geminal-atom catalysts (HF–Cu/GACs), simultaneously accelerating oxidative addition and suppressing β-hydride elimination, enabling broad-scope and highly selective C(sp2)–C(sp3) cross-coupling between aryl boronic esters and alkyl (pseudo)halides. These catalysts enable the synthesis of diverse pharmaceutically relevant intermediates in fewer steps, with higher yields and using safer, more sustainable conditions compared to traditional routes. Mechanistic studies reveal that the HF–Cu/GACs feature paired, low-valent Cu centers with minimal d-orbital holes, and that C–Br bond activation proceeds through a surface-mediated single-electron transfer between coadsorbed reactants, rather than free-radical rebound pathways. The findings here establish a generalizable strategy for electronic-state engineering of geminal metal sites to overcome long-standing challenges in cross-coupling chemistry and highlight the potential of heterogeneous Cu catalysts for the sustainable synthesis of fine chemicals and pharmaceuticals.
22
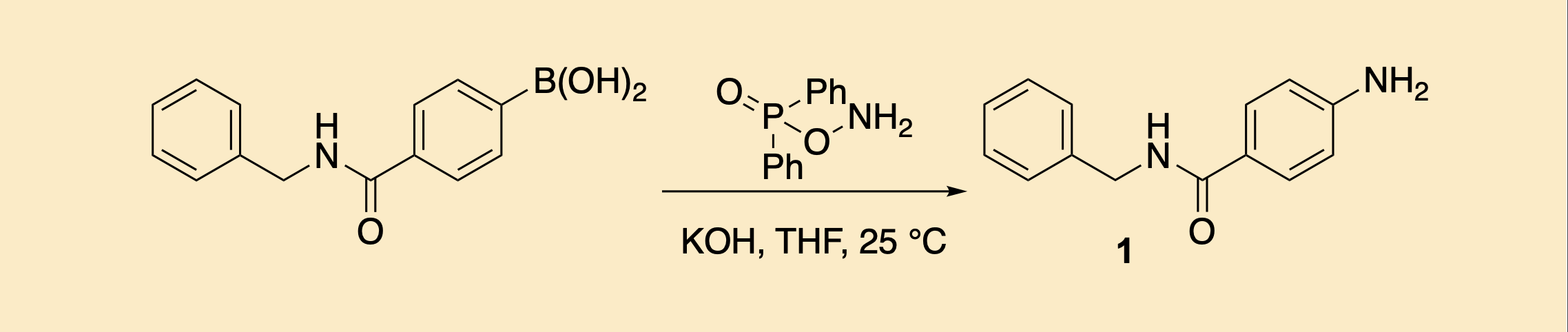
Amination of Aryl Boronic Acids Using O-(Diphenylphosphinyl)hydroxylamine (DPPH)
Kasie H. Leung and Richard Y. Liu
21
Ariel Y. Wang, Bayu I. Z. Ahmad, Carolyn Ma, Phillip J. Milner, Richard Y. Liu
Organic Photochemistry for Direct Light-Driven Separations
-
Compared to conventional methods, direct light-driven separations are promising strategies to achieve high selectivity while lowering energy cost. This perspective examines recent progress in photoswitch-enabled separations with a focus on concentration of CO2 and selective anion recovery from water. We highlight key design strategies for photo-pH-swing capture and identify current practical limitations toward translating solar-driven separations into technologies with meaningful impact.
20
Dual-Ligand System for Mild Decarbonylative Suzuki–Miyaura Cross-Coupling of Aroyl Chlorides
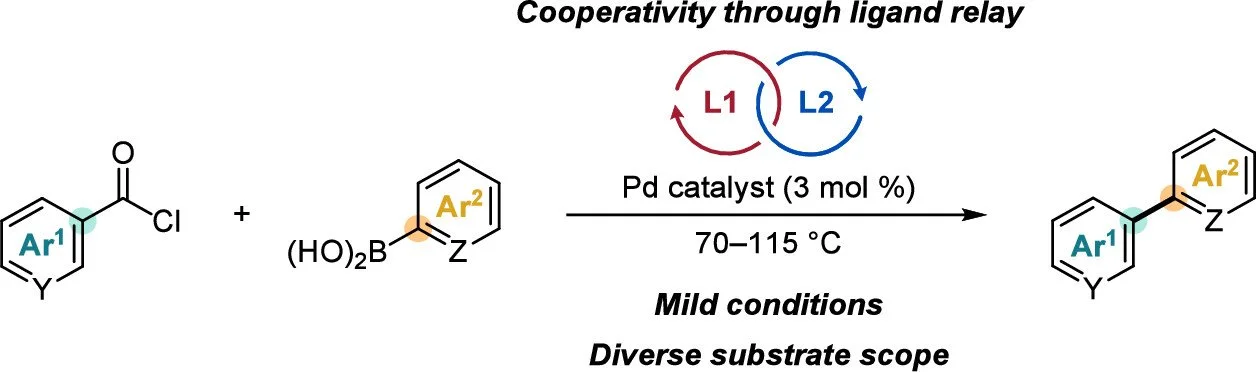
Marcus H. Sak, Yichen Jiang, Eric N. Jacobsen, Richard Y. Liu
-
Cooperativity between a pair of phosphine ligands enables general Pd-catalyzed decarbonylative Suzuki–Miyaura cross-couplings between (hetero)aroyl chlorides and (hetero)arylboronic acids under mild conditions. Experimental and computational studies support a ligand-relay mechanism in which each phosphine preferentially promotes different elementary steps, enhancing the yield and selectivity relative to using either ligand alone. These results validate empirical, mechanism-agnostic screening through pooling–deconvolution as a means for identifying synthetically enabling catalytic methods and mechanisms for multiligand cooperativity.
2025
19
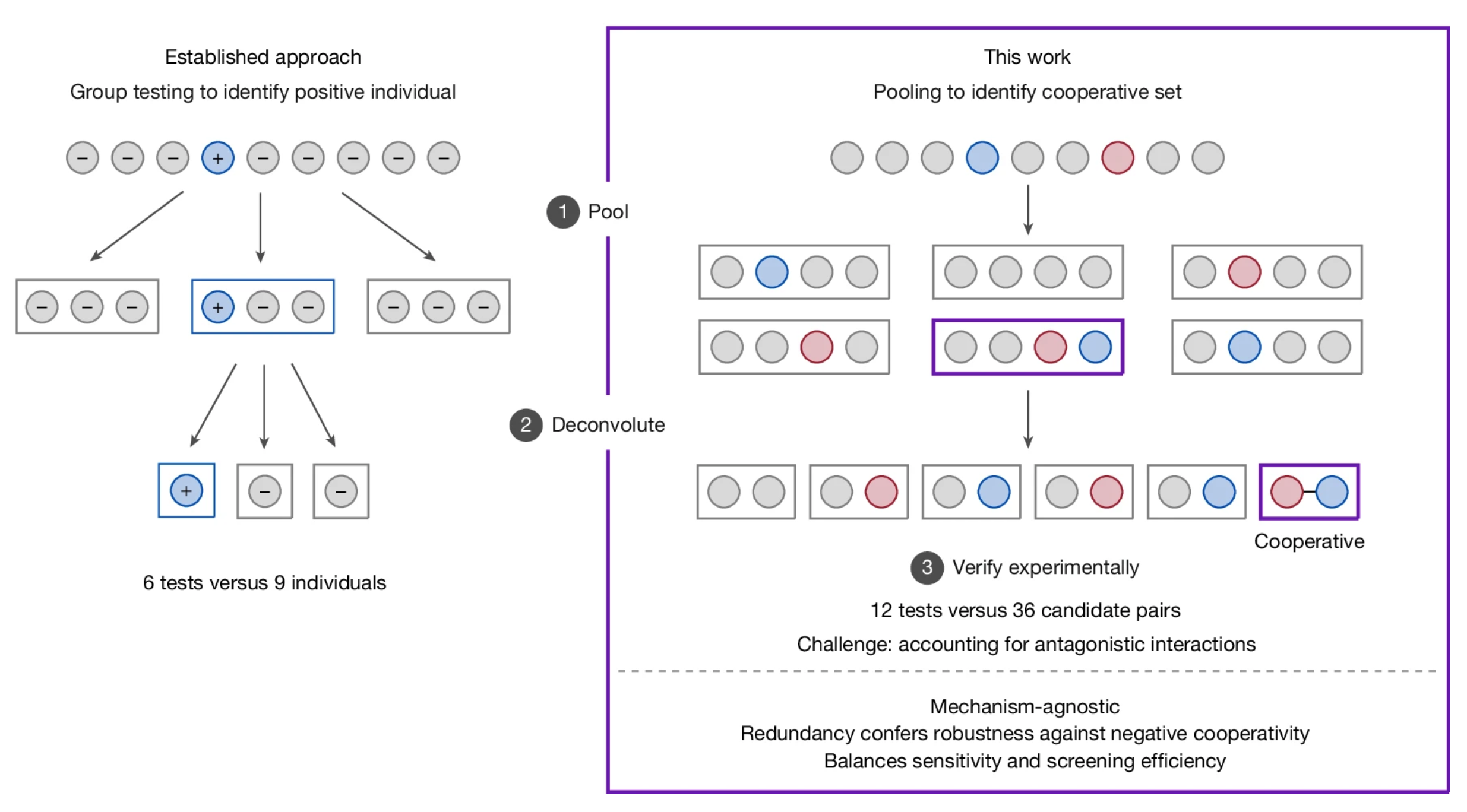
Accelerating the Discovery of Multicatalytic Cooperativity
Marcus H. Sak, Richard Y. Liu, Eugene E. Kwan, Eric N. Jacobsen
-
Cooperative catalysis, in which multiple catalytic units operate synergistically, underpins a variety of synthetically and mechanistically important organic reactions1,2,3,4. Despite its potential utility in new reactivity contexts, approaches to the discovery of cooperative catalysts have been limited, typically relying on serendipity or on previous knowledge of single-catalyst reactivity1,5. Systematic searches for unanticipated types of catalyst cooperativity must contend with vast combinatorial complexity and are therefore not undertaken6,7,8,9,10. Here we describe a pooling–deconvolution algorithm, inspired by group testing11, which identifies cooperative catalyst behaviours with low experimental cost while accommodating potential inhibitory effects between catalyst candidates. The workflow was validated first on simulated cooperativity data and then by experimentally identifying previously documented cooperativity between organocatalysts in an enantioselective oxetane-opening reaction. The workflow was then applied in a discovery context to a Pd-catalysed decarbonylative cross-coupling reaction, enabling the identification of several ligand pairs that promote the target transformation at substantially lower catalyst loading and temperature than previously reported with single-ligand systems.
18
Aminative Suzuki–Miyaura Coupling: Tackling Alkyl Nucleophiles and Addressing the Viability of the Electrophile-First Mechanism

Sophia Z. Li, Richard Y. Liu
-
Insertive cross-coupling reactions provide the option of repurposing widely available coupling partners for the formation of new linkages. By confronting a major limitation of the aminative Suzuki–Miyaura methods we recently reported, we achieve a general method for the Pd-catalyzed aminative coupling of primary and secondary alkyl boronic esters with aryl (pseudo)halides. Introducing a formal nitrene insertion into this Suzuki–Miyaura reaction diverts the outcome from the traditional C(sp2)─C(sp3) products to the C(sp2)─NH─C(sp3) analogues (N-aryl anilines). DFT calculations and experimental mechanistic studies indicate that C─N bond formation from the electrophilic aryl component occurs first, providing further evidence for this previously hypothetical pathway. By comparison of several transition state structures, we find that C─N bond formation likely takes place through an unusual dyotropic rearrangement of a LPd(Ar)NHX complex (X = OPOPh2).
17
Ion Pairing Enhances Hydroquinone Stability Toward Oxygen in Aqueous Electrochemical Carbon Dioxide Capture
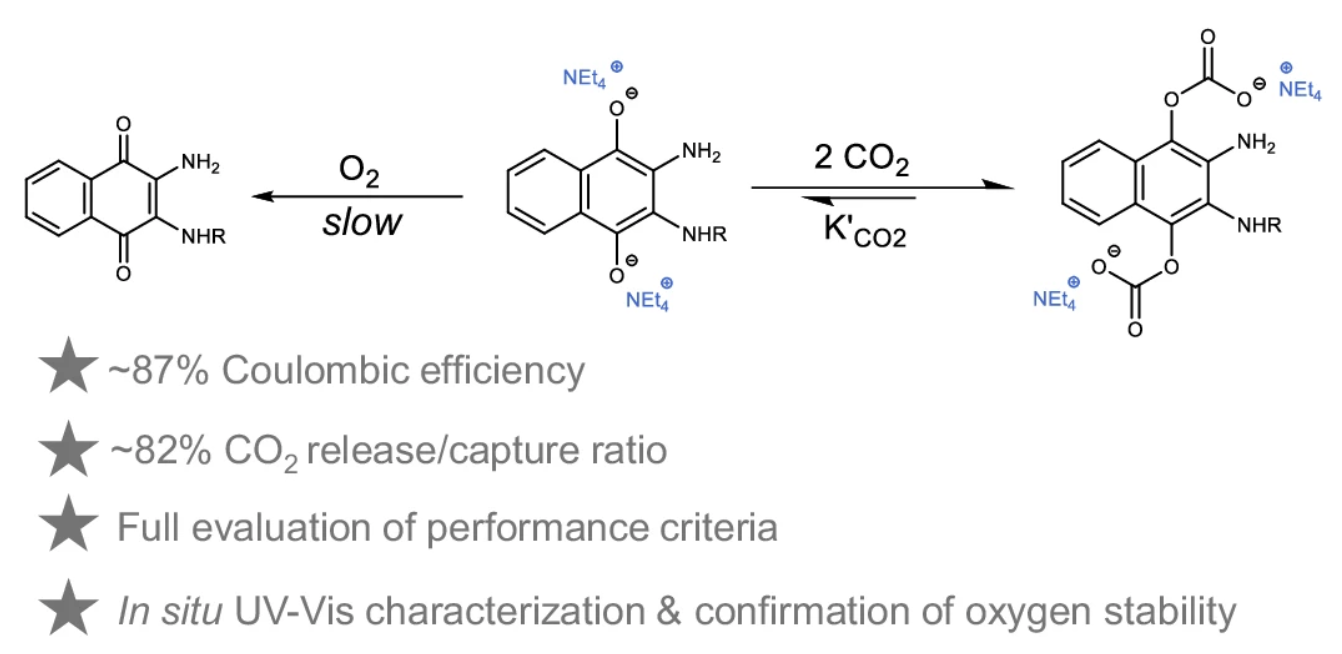
Abdulrahman M. Alfaraidi, Nina Ni, Jordan Sosa, Sara Lia, Mayar Alhelaili, Nathan H. Faialaga, Nawal M. Alghoraibi, Husain H. Al Naji, Ammar H. Alahmed, Aqil Jamal, Michael J. Aziz, Richard Y. Liu
-
The use of redox-active organic molecules for aqueous electrochemical carbon dioxide capture is limited by their tendency to undergo reversible oxidation by oxygen. Here we show that a naphthoquinone derivative, when reduced in the presence of tetraalkylammonium countercations, displays enhanced stability toward oxygen while maintaining carbon dioxide binding ability. By combining structural modification with control of non-covalent interactions, we mitigate a previously observed trade-off between carbon dioxide capture performance and resistance to aerobic oxidation. In situ spectrophotometry and comparative voltammetry indicate that ion pairing stabilizes the reduced quinone both by shifting its redox potential and by promoting carbon dioxide adduct formation. Among the cations tested, tetraethylammonium provides the most favorable balance, supporting efficient capture and release cycle with 87 % Coulombic efficiency and an energy cost of 157 kilojoules per mole of carbon dioxide from a gas mixture containing carbon dioxide, oxygen, and nitrogen. These findings illustrate how molecular design combined with electrolyte engineering can improve the durability of aqueous quinone-based electrochemical carbon capture systems and may inform the development of more robust and energy-efficient approaches for sustainable carbon management.
16
Pd-Catalyzed Azidation of Aryl (Pseudo) Halides
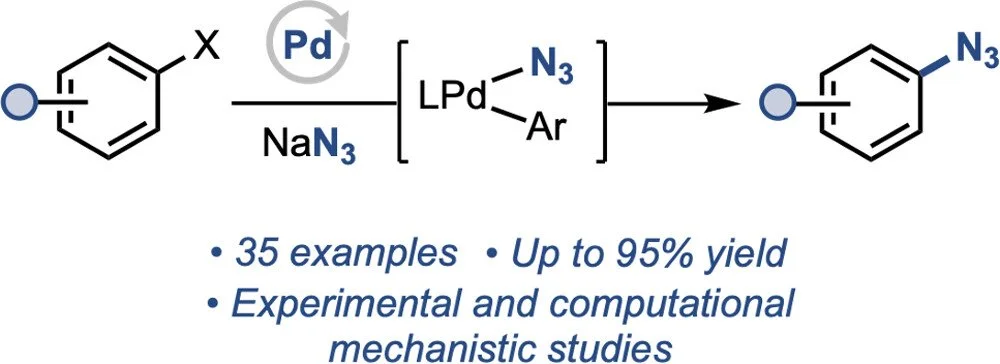
Shangyu Li, Polpum Onnuch, Richard Y. Liu
-
Organic azides are powerful tools in organic synthesis, chemical biology, and drug discovery. Pd catalysts have become general tools for C–N bond formation from aryl electrophiles, but azide stands out as a rare example of a nitrogen-centered nucleophile for which suitable catalysts have not been identified. Herein, we report the development of an effective Pd-catalyzed method for the synthesis of aryl azides from aryl bromides and aryl triflates using sodium azide as a convenient reagent. A variety of heterocycles, as well as aryl electrophiles incompatible with existing approaches (e.g., triflates), undergo efficient azidation. Experimental and computational mechanistic studies point to three distinct roles of the bulky ancillary phosphine ligands critical to facilitating this reaction. First, the structure of the phosphine lowers the barrier to reductive elimination from LPd(Ar)N3; second, it minimizes the formation of off-cycle Pd2(μ-N3)2 dimer; finally, it affords resistance of the LPd(0) toward inactivation by the aryl azide product.
15
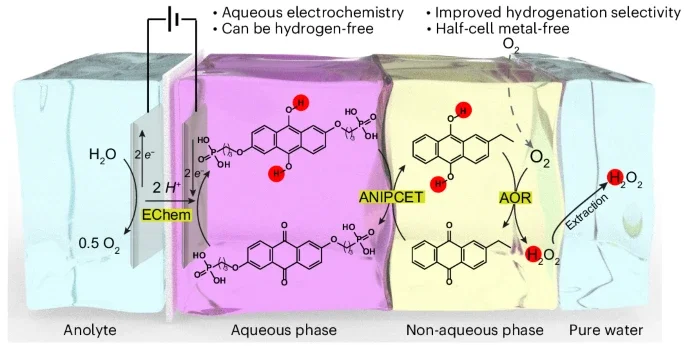
Electrifying Industrial Hydrogen Peroxide Production via Soft Interfacial Molecular Mediation
Dawei Xi, Yuheng Wu, Yuli Li, Zifei Yan, Richard Y. Liu, Michael J. Aziz
-
Hydrogen peroxide is manufactured industrially via the anthraquinone autoxidation process—a typical thermocatalytic non-aqueous method. Despite a high interest in using renewable electricity to drive such processes, electrifying non-aqueous syntheses remains a substantial challenge. Here we present a multi-phase electrochemical anthraquinone autoxidation process that leverages an aqueous–non-aqueous interfacial proton-coupled electron transfer method facilitated by heterogeneous molecular mediation. This design enables the reduction of aqueous anthraquinones with high efficiency at high current densities, using only carbon electrodes. The method operates with high selectivity through a quinhydrone intermediate and prevents the over-reduction of aromatics during thermocatalytic hydrogenation. This approach combines the benefits of aqueous electrochemistry with those of the traditional non-aqueous process to achieve high current density electrochemistry with rapid kinetics and mass transport, while avoiding unwanted electrolyte in the hydrogen peroxide product. This strategy bridges aqueous electrochemistry with non-aqueous chemistry and establishes a framework for the electrification and decentralization of other non-aqueous chemical processes.
14
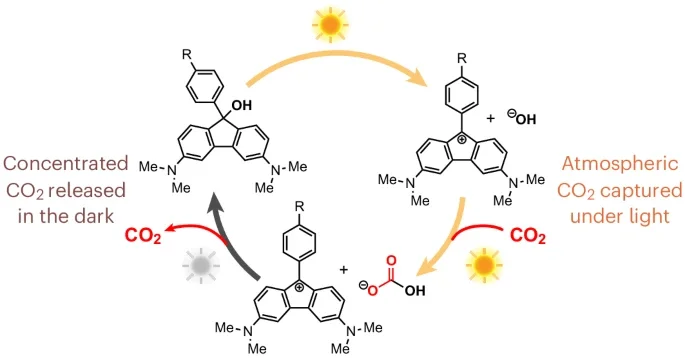
Reversible Fluorenol Photobases that Perform CO2 Capture and Concentration from Ambient Air
Michael Purdy, Ariel Y. Wang, Matthew C. Drummer, Daniel G. Nocera, Richard Y. Liu
-
Leading strategies for the capture of CO2 from point sources and directly from the atmosphere face high energy costs for thermal sorbent regeneration. Photochemical processes, driven by sunlight as the sole external stimulus, offer a promising alternative. Despite many reported examples of light-induced pH swings using metastable photoacids, the complementary mode of operation, using photoswitchable bases, has not been extensively considered. This is due in part to the rarity of photobases that can support large, reversible pH jumps in water. Here we report the design of fluorenol-based Arrhenius photobases that take advantage of excited-state aromaticity and ground-state antiaromaticity to generate large basicity swings with high reversibility. The system is oxygen stable, can be driven by natural sunlight and captures/concentrates CO2 from ambient air. We elucidated the mechanism of C–O dissociation using transient absorption spectroscopy to understand the high efficiency of hydroxide release. This study provides a framework for the design of photoreversible aqueous bases and guiding principles for their use in solar-powered CO2 management.
13
Synthesis of Diaryl Ethers by Formal Oxygen Insertion Between Suzuki–Miyaura Coupling Partners
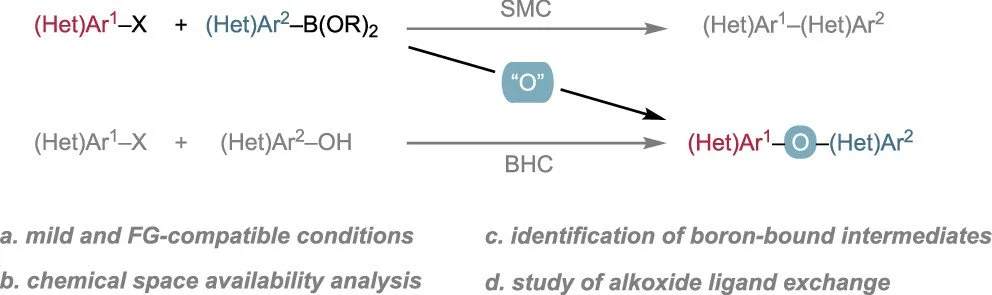
Yichen Jiang, Eugene E. Kwan, Yifan Ping, Richard Y. Liu
-
Because of the broad availability of Suzuki–Miyaura cross-coupling partners (aryl (pseudo)halides and organoboron reagents), methods that can repurpose them for the divergent synthesis of many compound classes are attractive. We report a convenient three-component coupling strategy that formally inserts an oxygen atom into a Suzuki–Miyaura transformation to yield diaryl ethers. The reaction, involving in situ generation of an aryl borate intermediate by oxidation, provides a disconnection complementary to that of traditional C–O cross-coupling. The method is effective on aryl chlorides, tolerates a broad range of functional groups, and can be used when the corresponding (hetero)aryl alcohol is less accessible (unavailable, unstable, or not easily synthesized). Mechanistic studies by 1H and 11B NMR spectroscopy reveal a critical and synergistic interplay between the Pd-independent and Pd-catalyzed steps. The formation of adducts between N-methylmorpholine N-oxide (NMO) and aryl borates may hinder oxidation. On the other hand, the byproduct N-methylmorpholine accelerates ligand exchange of LPd(Ar)X with Ar′O–B(OR)2.
12
Ni-Catalyzed Reductive Ring Contraction via Desulfurative Cross-Coupling
Jianhan Zhou, Richard Y. Liu
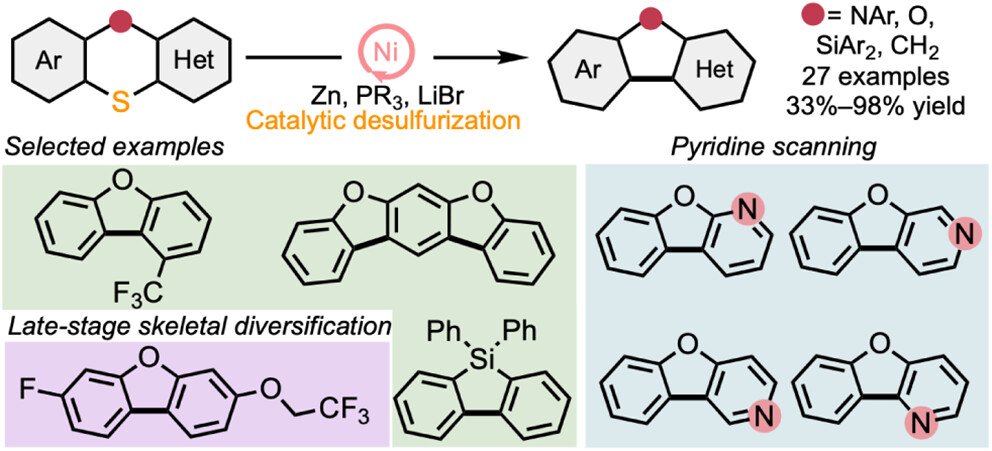
-
The construction of C–C bonds by contractive “deletion” of the sulfur atom from a C–S–C motif is a useful transformation for the synthesis of complex heterocycles. The transformation allows a user to take advantage of the nucleophilic reactivity of sulfur (e.g., for SNAr reactions) and remove that temporary atom at a late stage. A few homogeneous metal systems have been reported to mediate such desulfurizations, but under harsh conditions and, due to the formation of stable oligomeric sulfido complexes, without catalytic turnover. Here, we report a catalytic solution using an accessible Ni precatalyst, inexpensive additives, and mild conditions. The method provides rapid access to a broad scope of fused heterocycles. Preliminary mechanistic studies provide insight into key aspects of the transformation, including the order of C–S bond activations, the synergistic effects of the unusual combination of additives, and the fate of the excised sulfur atom.
11
Photochemical CO Release from a Bench-Stable, Recyclable Organic Platform: Applications to Carbonylative Cross-Coupling
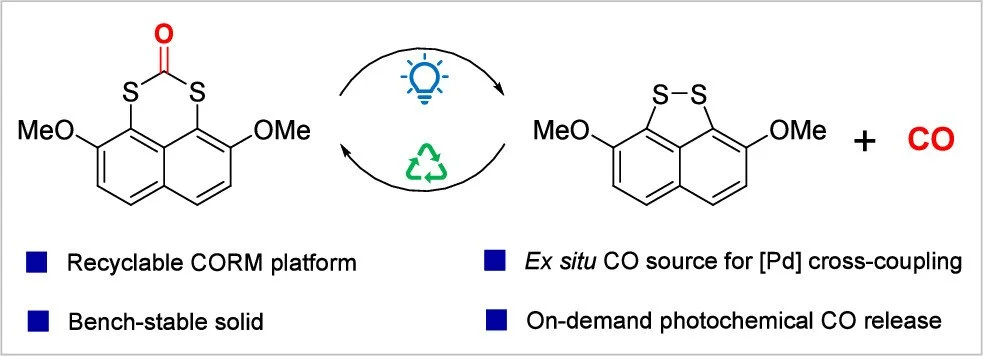
Khashayar Rajabimoghadam, Nathan H. Faialaga, Naoki Naito, Yuli Li, Richard Y. Liu
-
1,8-Naphthylene dithiocarbonates enable the safe and light-controlled release of carbon monoxide gas (CO). The disulfide byproducts, produced upon blue-light irradiation of the dithiocarbonates, were readily recovered and recycled by reduction and reaction with 1,1-carbonyldiimidazole (CDI). The photochemical release process was investigated by 1H NMR, GC, FTIR, UV–vis, and DFT calculations. The ex situ evolution of CO from this recyclable platform was applied to a range of Pd-catalyzed carbonylative cross-coupling reactions using a two-chamber glass apparatus.
10
‘Ring-Walking’ Aryl Cross-Coupling Reactions Involving Palladium Aryne Intermediates
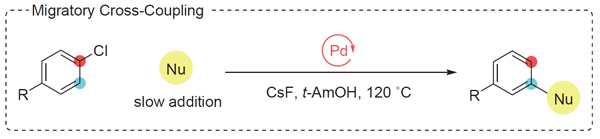
Eva A. Bayer, Yoshiya Sekiguchi, Polpum Onnuch, Richard Y. Liu
-
The regiospecificity of conventional cross-coupling reactions, though advantageous for its predictability and retrosynthetic simplicity, constrains chemical-space exploration. Discovery efforts have become biased toward examining substitution patterns for which coupling partners are readily obtainable. To address this problem, we have developed a migratory (‘ring-walking’) cross-coupling approach that integrates the isomerization of arylpalladium(II) intermediates into catalytic cycles. A reversible isomerization was achieved through ligand design and the use of cesium fluoride as the base, and this process was then combined with a dynamic kinetic resolution of the regioisomeric aryl halides with a broad range of oxygen- and nitrogen-centered nucleophiles. The method permits rapid access to meta-substituted arenes from para-substituted electrophiles. This account summarizes the key mechanistic principles established during the development of these reactions.
9
Discovery of a Fluorinated Macrobicyclic Antibiotic Through Chemical Synthesis
Ben I. C. Tresco, Kelvin J. Y. Wu, Antonio Ramkissoon, Elena V. Aleksandrova, Michael Purdy, Dominic N. Y. See, Richard Y. Liu, Yury S. Polikanov, Andrew G. Myers
-
The emergence of bacterial antimicrobial resistance threatens to undermine the utility of antibiotic therapy in medicine. This threat can be addressed, in part, by reinventing existing antibiotic classes using chemical synthesis. Here we present the discovery of BT-33, a fluorinated macrobicyclic oxepanoprolinamide antibiotic with broad-spectrum activity against multidrug-resistant bacterial pathogens. Structure–activity relationships within the macrobicyclic substructure reveal structural features that are essential to the enhanced potency of BT-33 as well as its increased metabolic stability relative to its predecessors clindamycin, iboxamycin and cresomycin. Using X-ray crystallography, we determine the structure of BT-33 in complex with the bacterial ribosome revealing that its fluorine atom makes an additional van der Waals contact with nucleobase G2505. Through variable-temperature 1H NMR experiments, density functional theory calculations and vibrational circular dichroism spectroscopy, we compare macrobicyclic homologues of BT-33 and a C7 desmethyl analogue and find that the C7 methyl group of BT-33 rigidifies the macrocyclic ring in a conformation that is highly preorganized for ribosomal binding.
8
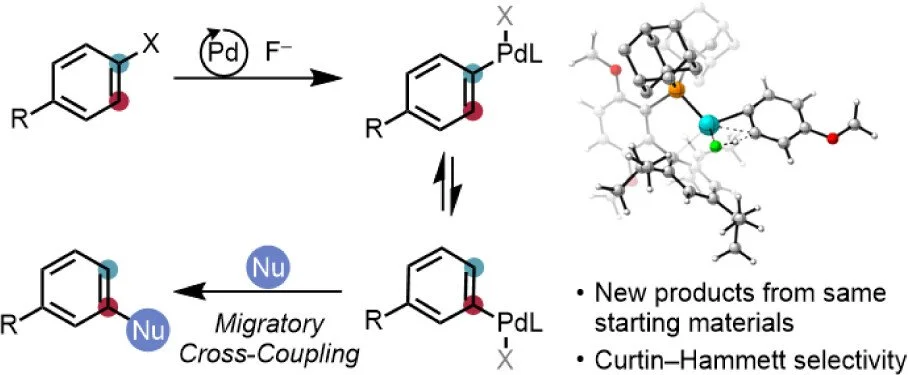
Migratory Aryl Cross-Coupling
Yoshiya Sekiguchi, Polpum Onnuch, Yuli Li, Richard Y. Liu
-
A fundamental property of cross-coupling reactions is regiospecificity, meaning that the site of bond formation is determined by the leaving group’s location on the electrophile. Typically, achieving a different substitution pattern requires the synthesis of a new, corresponding starting-material isomer. As an alternative, we proposed the development of cross-coupling variants that would afford access to multiple structural isomers from the same coupling partners. Here, we first demonstrate that a bulky palladium catalyst can facilitate the efficient, reversible transposition of aryl halides by temporarily forming metal aryne species. Despite the nearly thermoneutral equilibrium governing this process, combining it with the gradual addition of a suitable nucleophile results in dynamic kinetic resolution of the isomeric intermediates and high yields of unconventional product isomers. The method accommodates a range of oxygen- and nitrogen-centered nucleophiles and tolerates numerous common functional groups. A Curtin–Hammett kinetic scheme is supported by computational and experimental data, providing a general mechanistic framework for extending this migratory cross-coupling concept.
7
Synthesis of Arenesulfenyl Fluorides and Fluorosulfenylation of Alkenes, Alkynes, and a-Diazocarbonyl Compounds
Nathan H. Faialaga, Dana P. Gephart, Breno D. Silva, Richard Y. Liu

-
Sulfenyl fluorides are organic compounds of sulfur in formal oxidation state +2 with the formula R−S−F. Although the chloride, bromide, and iodide analogues have been extensively described in the literature, arenesulfenyl fluorides remain essentially unstudied. These structures have been implicated as putative intermediates in established processes to access polyfluorinated sulfur species; however, definitive and direct evidence of their existence has not been obtained, nor has a systematic understanding of their reactivity. Here, we report the synthesis, isolation, and spectroscopic characterization of several arenesulfenyl fluorides, including structural analysis of 2,4-dinitrobenzenesulfenyl fluoride and 4-cyano-2-nitrobenzenesulfenyl fluoride by single-crystal X-ray diffraction. The functional group undergoes direct, efficient, and highly regioselective anti-addition to alkenes and alkynes, as well as insertion by carbenes. The resulting α- or β-fluoro thioether adducts can be readily transformed into useful fluorinated motifs, for example by modification of the sulfur groups (to sulfonamides or sulfonyl fluorides), by sulfur elimination (to generate formal C−H fluorination products), or by Julia–Kocienski olefination (to form vinyl fluorides). Thus, we establish that sulfenyl fluorides are unexpectedly accessible and stable compounds, which serve as versatile reagents for the production of fluorinated organic compounds.
2024
6
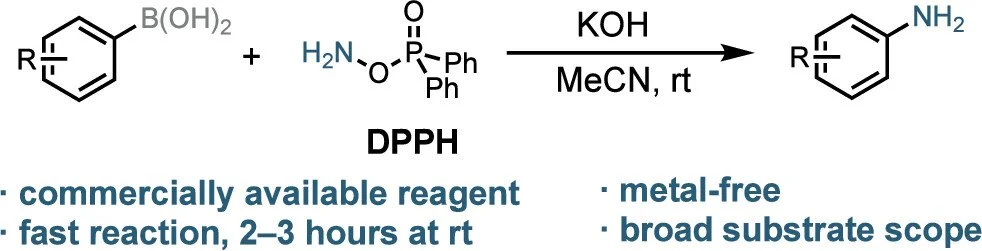
Rapid and General Amination of Aryl Boronic Acids and Esters Using O-(Diphenylphosphinyl)hydroxylamine (DPPH)
Matthew G. Kung, Polpum Onnuch and Richard Y. Liu
-
O-(Diphenylphosphinyl)hydroxylamine (DPPH) is a general reagent for the conversion of (hetero)aryl boronic acids and esters to primary anilines. The transformation proceeds rapidly at rt and exhibits a broad substrate scope and exceptional functional-group tolerance. In terms of rate, the reaction is relatively insensitive to the electronic properties of the substrate, in contrast to similar reactions using electrophilic amination reagents such as hydroxylamine-O-sulfonic acid. Consequently, this protocol is particularly useful for accessing electron-deficient (hetero)aryl anilines, which had been challenging to prepare using prior methods
5
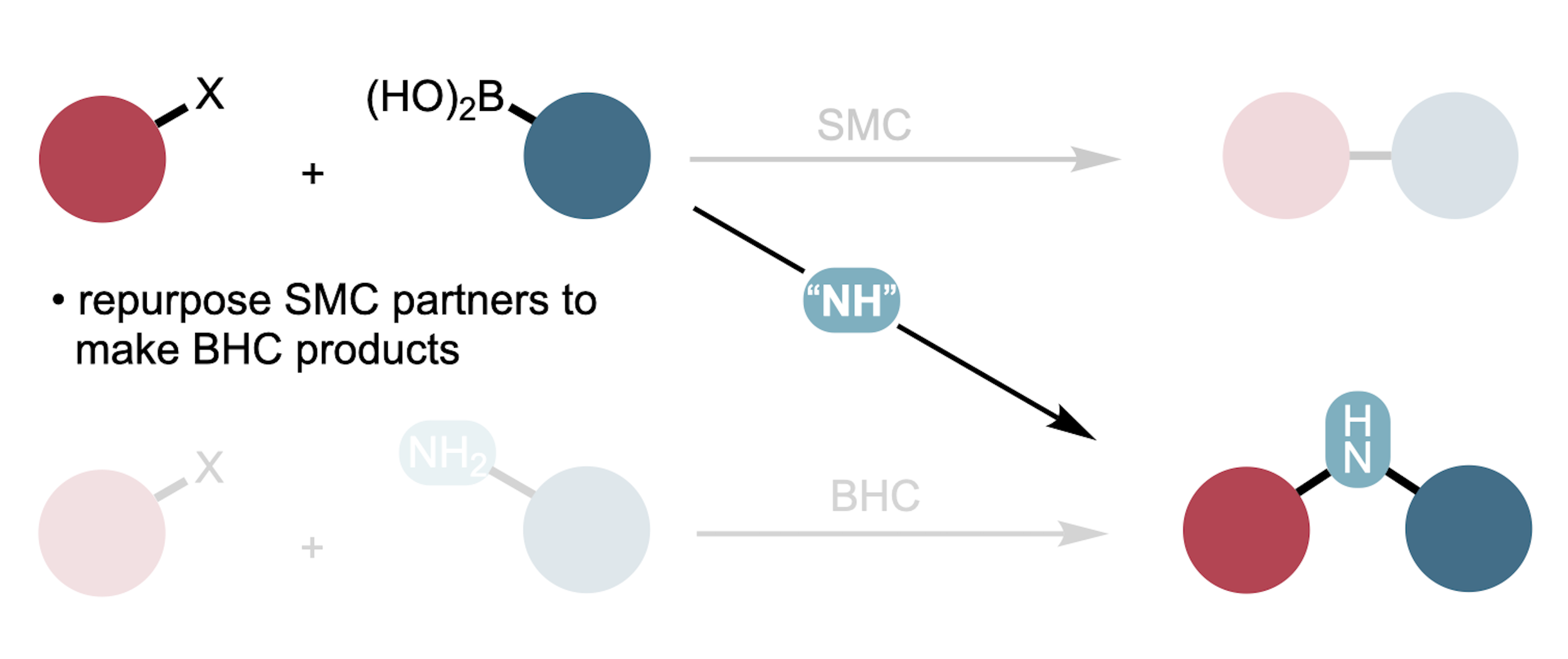
Aminative Suzuki–Miyaura Coupling
Polpum Onnuch, Kranthikumar Ramagonolla, Richard Y. Liu
-
The Suzuki–Miyaura and Buchwald–Hartwig coupling reactions are widely used to form carbon-carbon (C–C) and carbon-nitrogen (C–N) bonds, respectively. We report the incorporation of a formal nitrene insertion process into the Suzuki–Miyaura reaction, altering the products from C–C–linked biaryls to C–N–C–linked diaryl amines and thereby joining the Suzuki–Miyaura and Buchwald–Hartwig coupling pathways to the same starting-material classes. A combination of a bulky ancillary phosphine ligand on palladium and a commercially available amination reagent enables efficient reactivity across aryl halides and pseudohalides, boronic acids and esters, and many functional groups and heterocycles. Mechanistic insights reveal flexibility on the order of bond-forming events, suggesting potential for expansion of the aminative cross-coupling concept to encompass diverse nucleophiles and electrophiles as well as four-component variants.
4
Dawei Xi, Abdulrahman M. Alfaraidi, Jinxu Gao, Thomas Cochard, Luana C. I. Faria, Zheng Yang, Thomas Y. George, Taobo Wang, Roy G. Gordon, Richard Y. Liu, Michael J. Aziz
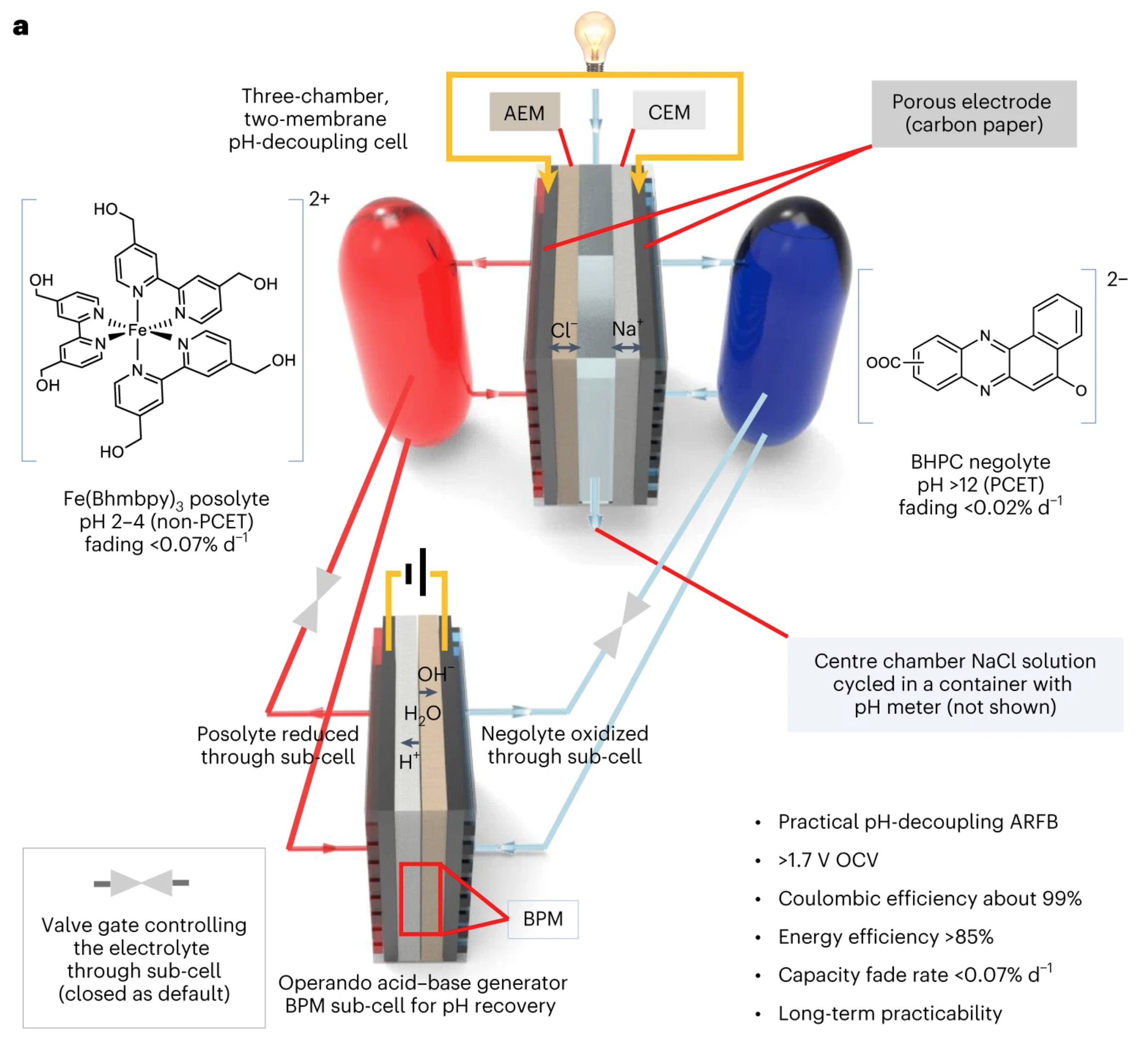
Mild pH-decoupling aqueous flow battery with practical pH recovery
-
Establishing a pH difference between the two electrolytes (pH decoupling) of an aqueous redox flow battery (ARFB) enables cell voltages exceeding the 1.23 V thermodynamic water-splitting window, but acid–base crossover penalizes efficiency and lifetime. Here we employ mildly acidic and mildly alkaline electrolytes to mitigate crossover, achieving high round-trip energy efficiency with open circuit voltage >1.7 V. We implemented an acid–base regeneration system to periodically restore electrolytes to their initial pH values. The combined system exhibited capacity fade rate <0.07% per day, round-trip energy efficiency >85% and approximately 99% Coulombic efficiency during stable operation for over a week. Cost analysis shows that the tolerance of acid–base crossover could be increased if the pH-decoupling ARFB achieved a higher voltage output and lower resistance. This work demonstrates principles for improving lifespan, rate capability and energy efficiency in high-voltage pH-decoupling ARFBs and pH recovery concepts applicable for pH-decoupling systems.
3
Kelvin J. Y. Wu, Ben I. C. Tresco, Antonio Ramkissoon, Elena V. Aleksandrova, Egor A. Syroegin, Dominic N. Y. See, Priscilla Liow, Georgia A. Dittemore, Meiyi yu, Giambattisa Testolin, Matthew J. Mitcheltree, Richard Y. Liu, Maxim S. Svetlov, Yury S. Polikanov, Andrew G. Myers
An Antibiotic Reorganized for Ribosomal Binding Overcomes Antimicrobial Resistance
-
We report the design conception, chemical synthesis, and microbiological evaluation of the bridged macrobicyclic antibiotic cresomycin (CRM), which overcomes evolutionarily diverse forms of antimicrobial resistance that render modern antibiotics ineffective. CRM exhibits in vitro and in vivo efficacy against both Gram-positive and Gram-negative bacteria, including multidrug-resistant strains of Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa. We show that CRM is highly preorganized for ribosomal binding by determining its density functional theory–calculated, solution-state, solid-state, and (wild-type) ribosome-bound structures, which all align identically within the macrobicyclic subunits. Lastly, we report two additional x-ray crystal structures of CRM in complex with bacterial ribosomes separately modified by the ribosomal RNA methylases, chloramphenicol-florfenicol resistance (Cfr) and erythromycin-resistance ribosomal RNA methylase (Erm), revealing concessive adjustments by the target and antibiotic that permit CRM to maintain binding where other antibiotics fail.
2023
An Extremely Stable and Soluble NH2-Substituted Anthraquinone Electrolyte for Aqueous Redox Flow Batteries
2
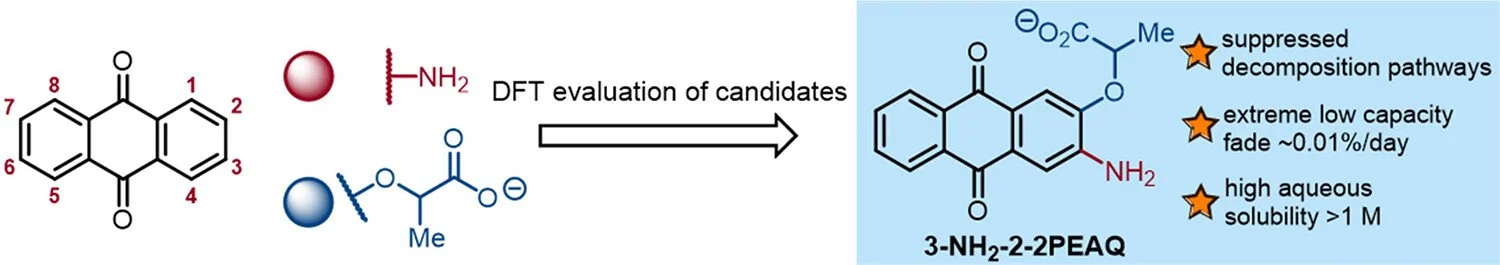
Abdulrahman M. Alfaraidi, Dawei Xi, Nina Ni, Thomas Y. George, Tatsuhiro Tsukamoto, Roy G. Gordon, Michael J. Aziz, Richard Y. Liu.
-
Description text goes hSeparation of carbon dioxide (CO2) from point sources or directly from the atmosphere can contribute crucially to climate change mitigation plans in the coming decades. A fundamental practical limitation for the current strategies is the considerable energy cost required to regenerate the sorbent and release the captured CO2 for storage or utilization. A directly photochemically driven system that demonstrates efficient passive capture and on-demand CO2 release triggered by sunlight as the sole external stimulus would provide an attractive alternative. However, little is known about the thermodynamic requirements for such a process or mechanisms for modulating the stability of CO2-derived dissolved species by using photoinduced metastable states. Here, we show that an organic photoswitchable molecule of precisely tuned effective acidity can repeatedly capture and release a near-stoichiometric quantity of CO2 according to dark–light cycles. The CO2-derived species rests as a solvent-separated ion pair, and key aspects of its excited-state dynamics that regulate the photorelease efficiency are characterized by transient absorption spectroscopy. The thermodynamic and kinetic concepts established herein will serve as guiding principles for the development of viable solar-powered negative emission technologies.ere
1
Reversible CO2 Capture and On-Demand Release by an Acidity-Matched Organic Photoswitch
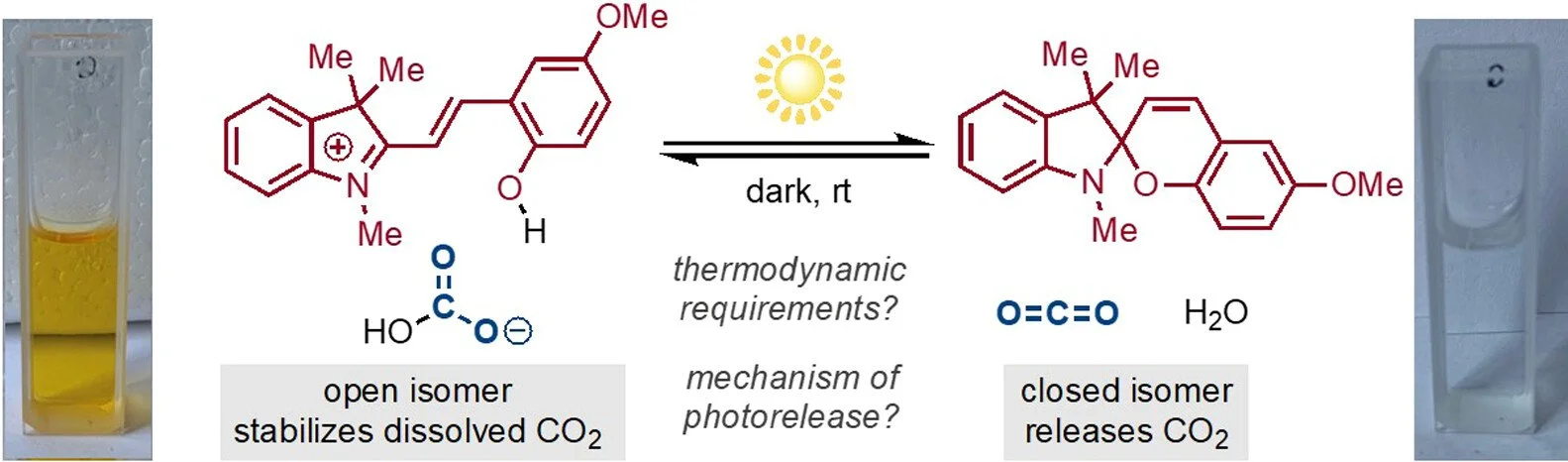
Abdulrahman M. Alfaraidi, Bryan Kudisch, Nina Ni, Jayden Thomas, Thomas Y. George, Khashayar Rajabimoghadam, Haihui Joy Jiang, Daniel G. Nocera, Michael J. Aziz, Richard Y. Liu.
-
Separation of carbon dioxide (CO2) from point sources or directly from the atmosphere can contribute crucially to climate change mitigation plans in the coming decades. A fundamental practical limitation for the current strategies is the considerable energy cost required to regenerate the sorbent and release the captured CO2 for storage or utilization. A directly photochemically driven system that demonstrates efficient passive capture and on-demand CO2 release triggered by sunlight as the sole external stimulus would provide an attractive alternative. However, little is known about the thermodynamic requirements for such a process or mechanisms for modulating the stability of CO2-derived dissolved species by using photoinduced metastable states. Here, we show that an organic photoswitchable molecule of precisely tuned effective acidity can repeatedly capture and release a near-stoichiometric quantity of CO2 according to dark–light cycles. The CO2-derived species rests as a solvent-separated ion pair, and key aspects of its excited-state dynamics that regulate the photorelease efficiency are characterized by transient absorption spectroscopy. The thermodynamic and kinetic concepts established herein will serve as guiding principles for the development of viable solar-powered negative emission technologies.ere

Prior Work
27. Richard Y. Liu, Shao-Xiong Lennon Luo, Elizabeth S. Hirst, Christopher J. Doona, Timothy M. Swager. “Bifunctional Diazirine Reagent for Covalent Dyeing of Kevlar and Inert Polymer Materials.” Polym. Chem. 2023, 14, 4205-4215. (link)
26. Anthony J. Rojas, Justin M. Wolfe, Heemal H. Dhanjee, Ivan Buslov, Nicholas L. Truex, Richard Y. Liu, Walter Massefski, Bradley L. Pentelute, Stephen L. Buchwald. “Palladium–Peptide Oxidative Addition Complexes for Bioconjugation.” Chem. Sci. 2022, 13, 11891–11895. (link)
25. Richard Y. Liu*, Sheng Guo*, Shao-Xiong Lennon Luo, Timothy M. Swager. “Solution-Processable Microporous Polymer Platform for Heterogenization of Diverse Photoredox Catalysts.” Nat. Commun. 2022, 13, 1–8. (* denotes equal contribution.) (link)
24. Máté J. Bezdek, Shao-Xiong Lennon Luo, Richard Y. Liu, Qilin He, Timothy M. Swager. “Trace Hydrogen Sulfide Sensing Inspired by Polyoxometalate-Mediated Aerobic Oxidation.” ACS Cent. Sci. 2021, 7, 1572–1580. (link)
23. Shao-Xiong Lennon Luo, Richard Y. Liu, Sungsik Lee, Timothy M. Swager. “Electrocatalytic Isoxazoline–Nanocarbon Metal Complexes.” J. Am. Chem. Soc. 2021, 143, 10441–10453. (link)
22. Michael W. Gribble, Jr.‡, Richard Y. Liu‡, Stephen L. Buchwald. “Evidence for Simultaneous Dearomatization of Two Aromatic Rings under Mild Conditions in Cu(I)-Catalyzed Direct Asymmetric Dearomatization of Pyridine.” J. Am. Chem. Soc. 2020, 142, 11252–11269. (‡ denotes corresponding author.) (link)
21. Richard Y. Liu, Stephen L. Buchwald. “CuH-Catalyzed Hydrofunctionalization: From Hydroamination to Carbonyl Addition.” Acc. Chem. Res. 2020, 53, 1229–1243. (link)
20. Richard Y. Liu*, Joseph M. Dennis*, Stephen L. Buchwald. “The Quest for the Ideal Base: Rational Design of a Nickel Precatalyst Enables Mild, Homogeneous C–N Cross-Coupling.” J. Am. Chem. Soc. 2020, 9, 4500–4507. (link)
19. Chengxi Li*, Kwangmin Shin*, Richard Y. Liu, Stephen L. Buchwald. “Engaging Aldehydes in CuH-Catalyzed Reductive Coupling Reactions: Stereoselective Allylation from 1,3-Diene Pronucleophiles.”
Angew. Chem. Int. Ed. 2020, 58, 17074–17080. (* denotes equal contribution.) (link)
18. Jessica Xu, Richard Y. Liu, Charles S. Yeung, Stephen L. Buchwald. “Monophosphine Ligands Promote Pd-Catalyzed C–S Cross-Coupling Reactions at Room Temperature with Soluble Bases.” ACS Catal. 2019, 9, 6461–6466. (link)
17. Chengxi Li, Richard Y. Liu, Luke T. Jesikiewicz, Yang Yang, Peng Liu, Stephen L. Buchwald. “CuH-Catalyzed Regio- and Enantioselective Ketone Allylation with 1,3-Dienes: Scope, Mechanism, and Applications.” J. Am. Chem. Soc. 2019, 141, 5062–5070. (link)
16. Joseph M. Dennis, Nicholas A. White, Richard Y. Liu, Stephen L. Buchwald. “Pd-Catalyzed C–N Coupling Reactions Facilitated by Amine Bases: Mechanistic Investigation Leads to Enhanced Reactivity in the Arylation of Weakly Binding Amines.” ACS Catal. 2019, 9, 3822–3830. (link)
15. Richard Y. Liu*, Yujing Zhou*, Yang Yang, Stephen L. Buchwald. “Enantioselective Allylation from Allene, a Petroleum Cracking Byproduct.” J. Am. Chem. Soc. 2019, 141, 2251–2256. (* denotes equal contribution.) (link)
14. Joseph M. Dennis*, Nicholas A. White*, Richard Y. Liu, Stephen L. Buchwald. “Breaking the Base Barrier: An Electron-Deficient Palladium Catalyst Enables the Use of Amine Bases in C–N Coupling.” J. Am. Chem. Soc. 2018, 140, 4721–4725. (* denotes equal contribution.) (link)
13. Richard Y. Liu and Stephen L. Buchwald. “Copper-Catalyzed Enantioselective Hydroamination of Alkenes.” Org. Synth. 2018, 95, 80–96. (Invited submission to Organic Syntheses.) (link)
12. Erica Y. Tsai, Richard Y. Liu, Yang Yang, Stephen L. Buchwald. “A Regio- and Enantioselective CuH-Catalyzed Ketone Allylation with Terminal Allenes.” J. Am. Chem. Soc. 2018, 140, 2007–2011. (link)
11. Richard Y. Liu*, Minwoo Bae*, Stephen L. Buchwald. “Mechanistic Insight Facilitates Discovery of a Mild and Efficient Copper-Catalyzed Dehydration of Primary Amides to Nitriles Using Hydrosilanes.” J. Am. Chem. Soc. 2018, 140, 1627–1631. (* denotes equal contribution.) (link)
10. Yujing Zhou, Jeffrey S. Bandar, Richard Y. Liu, Stephen L. Buchwald. “CuH-Catalyzed Asymmetric Reduction of α,β-Unsaturated Carboxylic Acids to β-Chiral Aldehydes.” J. Am. Chem. Soc. 2018, 140, 606–609. (link)
9. Gang Lu, Richard Y. Liu, Yang Yang, Cheng Fang, Daniel S. Lambrecht, Stephen L. Buchwald, Peng Liu. “Ligand–Substrate Dispersion Facilitates the Copper-Catalyzed Hydroamination of Unactivated Olefins.” J. Am. Chem. Soc. 2017, 139, 16548–16555. (link)
8. Yongho Park, Kaid C. Harper, Nadine Kuhl, Eugene E. Kwan, Richard Y. Liu, Eric N. Jacobsen. “Macrocyclic Bis-Thioureas Catalyze Stereospecific Glycosylation Reactions.” Science 2017, 355, 162–166. (link)
7. Michael W. Gribble, Jr., Michael T. Pirnot, Jeffrey S. Bandar, Richard Y. Liu, Stephen L. Buchwald. “Asymmetric Copper Hydride-Catalyzed Markovnikov Hydrosilylation of Vinylarenes and Vinyl Heterocycles.” J. Am. Chem. Soc. 2017, 139, 2192–2195. (link)
6. Richard Y. Liu, Yang Yang, Stephen L. Buchwald. “Regiodivergent and Diastereoselective CuH-Catalyzed Allylation of Imines with Terminal Allenes.” Angew. Chem. Int. Ed. 2016, 55, 14077–14080. (link)
5. Benjamin H. Rotstein, Lu Wang, Richard Y. Liu, Jon Patteson, Eugene E. Kwan, Neil Vasdev, Steven H. Liang. “Mechanistic Studies and Radiofluorination of Structurally Diverse Pharmaceuticals with Spirocyclic Iodonium(III) Ylides.” Chem. Sci. 2016, 7, 4407–4417. (link)
4. Eugene E. Kwan, Richard Y. Liu. “Enhancing NMR Prediction for Organic Compounds Using Molecular Dynamics.” J. Chem. Theory Comput. 2015, 11, 5083–5089. (link)
3. Richard Y. Liu, Masayuki Wasa, Eric N. Jacobsen. “Enantioselective Synthesis of Tertiary α-Chloro Esters by Non-Covalent Catalysis.” Tetrahedron Lett. 2014, 56, 3428–3430. (Invited submission in honor of Harry H. Wasserman.) (link)
2. Masayuki Wasa, Richard Y. Liu, Stéphane P. Roche, Eric N. Jacobsen. “Asymmetric Mannich Synthesis of α-Amino Esters by Anion-Binding Catalysis.” J. Am. Chem. Soc. 2014, 136, 12872–12875. (link)
1. Elisabeth T. Hennessey, Richard Y. Liu, Diana A. Iovan, Ryan A. Duncan, Theodore A. Betley. “Iron-Mediated Intermolecular N-Group Transfer Chemistry with Olefinic Substrates.” Chem. Sci. 2014, 5, 1526–1532. (link)